


RECLASSIFICATION OF MEDICAL DEVICES
COMPREHENSIVE ADJUSTMENT AND NOTIFICATION OF MEDICAL DEVICES IN ACCORDANCE WITH THE MDR CLASSIFICATION
GOFARM HELPS BRING NEW PRODUCTS TO MARKET AND ADAPT EXISTING PRODUCTS TO NEW REGULATIONS.

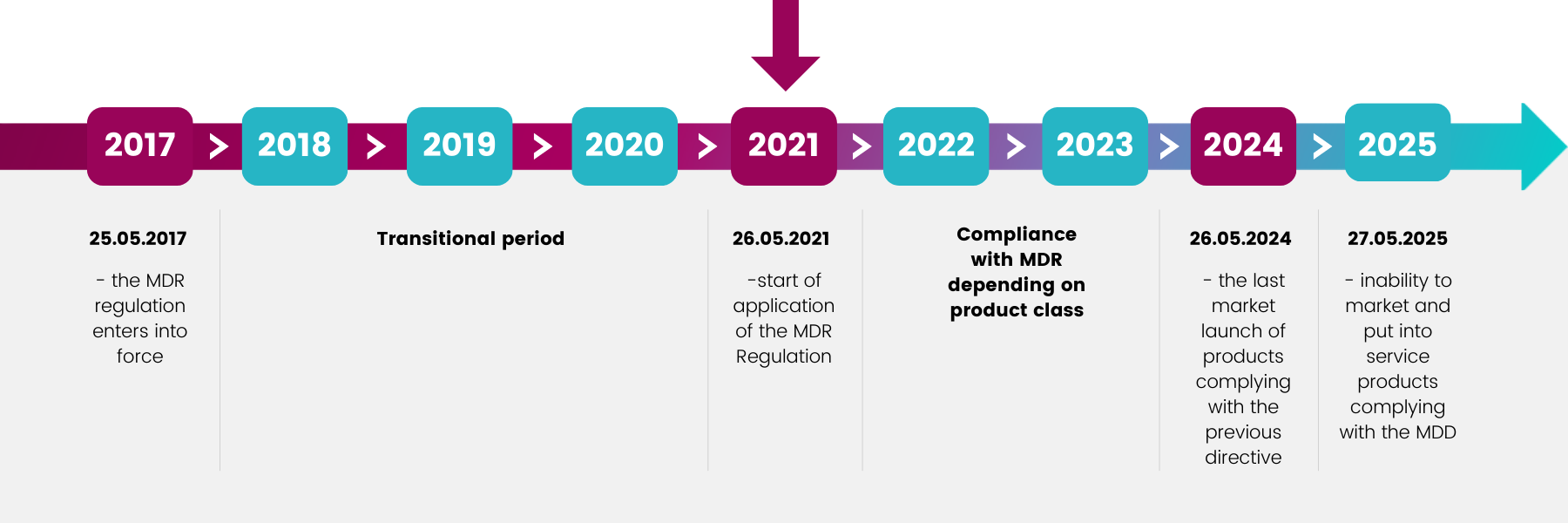
Due to the entry into force of the new Regulation MDR 2017/745,
the rules for the classification of medical devices are changing. Some items in your portfolio may need to be subjected to a procedure to adapt the product and its documentation to the new requirements.
The required procedures take time – don’t leave it for a later date.

REMEMBER THAT IT IS THE RESPONSIBILITY OF EVERY MANUFACTURER AND DISTRIBUTOR TO MAKE CERTAIN CHANGES TO THE MEDICAL DEVICE DOCUMENTATION.
ARE YOU READY FOR NEW REGULATIONS
TO KEEP YOUR PRODUCTS ON THE MARKET?
GET READY FOR CHANGE TODAY!
DON’T WAIT TILL THE VERY LAST MINUTE!
DO YOU HAVE PRODUCTS IN YOUR PORTFOLIO THAT REQUIRE RECLASSIFICATION?
SEE THE SCHEDULE OF CHANGES.
DID YOU KNOW…
Gofarm has successfully carried out
over 20 reclassifications of products
to higher grades in 16 months?
TRUST THE EXPERIENCE
AND KNOWLEDGE OF EXPERTS
WHO HAVE CARRIED OUT SUCH PROCEDURES
FOR THEIR CLIENTS
MANY TIMES BEFORE

")
CASE STUDY
Manufacturer X has a nasal spray that is currently a Class I medical device. The Manufacturer is wondering what to do to make sure that the product can continue to be sold after 26 May 2024.
According to the MDR, in order for such a medical device to remain on the market, its documentation must be adapted to the new requirements and in selected cases the device class must be changed from Class I to a higher class. Our company will conduct an audit of the product documentation and advise you on the steps to be taken and the deadline for introducing the changes in time. We will guide your company through the entire process and you will gain sales continuity.
")

COMPLEX ADAPTATION AND REGISTRATION OF PRODUCTS TO A HIGHER PRODUCT CLASS
- We will support you at every step
- We will carry out an audit of your product documentation and propose a schedule for the implementation of changes / advise which products require adjustment of technical documentation, certification, additional testing
- Rapid and efficient updating of documentation
- Save time
- Reliable, expert assistance from the Gofarm team
- Suggested notified body
- We will guide your company through the entire process and you will gain continuity of sale of your medical devices
PLEASE NOTE!
THE PROCESS OF BRINGING MEDICAL DEVICES INTO COMPLIANCE WITH THE MDR TAKES TIME AND OFTEN INVOLVES THE PARTICIPATION OF NOTIFIED BODIES.
TAKE THIS INTO ACCOUNT WHEN PLANNING TO IMPLEMENT CHANGES.
WE WILL HELP YOU
The aforementioned services are conducted on the basis of the following legal basis: Regulation (EU) 2017/745 of the European Parliament and of the Council of 5 April 2017 on medical devices, amending Directive 2001/83/EC, Regulation (EC) No 178/2002 and Regulation (EC) No 1223/2009 and repealing Council Directives 90/385/EEC and 93/42/EEC